|
One of the challenges of tissue
engineering, a promising cell-based
treatment for damaged or diseased
cartilage, is designing the scaffold
that provides structure while the
tissue regenerates. In addition to the
scaffold material's biocompatibility,
mechanical properties, and ease of
manufacturing, scaffold interactions
with the cells must also be considered.
In cartilage tissue engineering, a range
of scaffolds with various degrees of cell
attachment have been proposed, but
the attachment density and type have
yet to be optimized. Several techniques
have been developed to modulate cell
adhesion to the scaffold. These studies
suggest that the need for cell attachment
in cartilage tissue engineering may
vary with cell type, stage of differentiation,
culture condition, and scaffold
material. Further studies will elucidate
the role of cell attachment in cartilage
regeneration and enhance efforts
to engineer cell-based cartilage
therapies.
| HOW WOULD YOU... |
describe the overall significance
of this paper?
Tissue engineering is a promising
treatment for damaged or diseased
cartilage and usually requires a
scaffold to provide structure while
cells produce new cartilage matrix.
The scaffold supplies a substrate
for cell attachment to support
cell survival, differentiation, and
cartilage matrix deposition. In
cartilage tissue engineering, cell
adhesion to the scaffold via integrin
binding may vary, but the results
of these attachments are not fully
understood. Efforts to understand
the role of integrin binding and focal
adhesion formation in cartilage
tissue engineering could impact cell-based
therapies for cartilage repair.
describe this work to a
materials science and engineering
professional with no experience in
your technical specialty?
Cell adhesion to a tissue engineering
scaffold is an important regulator
of cell behavior and tissue
regeneration. Cell adhesion to
a material may be modulated by
treatments that improve wettability
or by grafting molecules that
provide sites for cell anchorage to
the surface. These techniques have
allowed for investigations into the
consequences of cell attachment on
cartilage tissue engineering, and are
expected to enhance cartilage repair.
describe this work to a
layperson?
In biological applications where
materials interact with cells, the
properties of the material that
control cell attachment are critically
important to cell function. Here,
we review the role of scaffolds as
a substrate for cell attachment
in the context of cartilage tissue
engineering. The effect of cell
attachment in cartilage regeneration
may vary with cell type, stage of
differentiation, culture condition and
scaffold material.
|
INTRODUCTION
Cartilage disease and injury are
substantial health issues; osteoarthritis
(OA) alone afflicts an estimated 15% of
the U.S. population (nearly 40 million
persons),1 and costs the U.S. economy
more than $60 billion per year.2 The
prevalence of OA increases rapidly with
age and with an aging U.S. population
it is expected that the incidence
and associated costs will increase
dramatically in the future.3 In spite of the magnitude of this problem, there
exist few adequate treatment options,
in part due to the fact that articular
cartilage exhibits limited capacity for
self-repair. Essentially no consistent
repair occurs in cartilage defects that
do not penetrate the subchondral bone.4
While full-thickness defects that do
penetrate the subchondral bone show
partial repair by bone marrow-derived
mesenchymal stromal cells (MSCs), the
repair tissue typically consists of a less
functional fibrocartilage, rather than
articular cartilage.5 Current therapies
for cartilage damage include abrasion
arthroplasty, microfracture, autologous
osteochondral transplantation, autologous
chondrocyte transplantation, and prosthetic
joint replacement;6 however, each
of these treatments has disappointing
limitations such as deficient long-term
repair, inadequate donor tissue/cell
availability, donor site morbidity, and
limited durability.
A promising treatment for damaged
or diseased cartilage is tissue
engineering, a technique that leverages
the principles of engineering and the
life sciences to develop substitutes
that restore, maintain, or improve the
function of tissues such as cartilage.7 A
successful tissue engineering solution
for cartilage repair will require a
combination of several components,
including the appropriate cell type;
biochemical and biomechanical signals
to encourage and maintain cell
metabolism and direct cell phenotype;
and a temporary artificial and/or
macromolecular scaffold to provide
structure for the regenerating tissue.8
The scaffold plays an important role
in maintaining cell function and
guiding tissue growth and has four
basic performance requirements:
three-dimensional and porous to allow nutrient and waste transport;
biocompatible and bioresorbable;
mechanical properties similar to the
native tissue; and appropriate cell
attachment that supports cell survival,
differentiation and matrix production.9
The focus of this article is on the
role of scaffolds as a substrate for
cell attachment in the context of
engineering cartilage tissue. Although
many of the challenges in the design
and manufacture of a scaffold for
tissue engineering are the same as
those for bio-inert medical implants,
tissue engineering scaffolds must
also be developed with cell-scaffold
interactions in mind, as they directly
regulate the biological function of
the cells. To engineer most tissues,
cell attachment sites are assumed to
be necessary features of the scaffold,
but in cartilage tissue engineering a
wide range of scaffolds with varying
levels of direct cell attachment are
regularly reported. See the sidebar for
a description of cartilage structure and
composition.
BIOLOGY OF CELL ATTACHMENT
| Cartilage Structure and Composition |
Articular cartilage is found on the ends of all diarthrodial joints
and is critical for joint motion.10 The ability to resist compression
and distribute loads allows cartilage to decrease the peak
stresses in subchondral bone.11 Cartilage is an aneural, avascular,
connective tissue composed mostly of extracellular matrix
(ECM) molecules and water. Approximately 7080% of cartilage
consists of water, which is vital for nutrient transfer and load
distribution. Chondrocytes, the cellular component of cartilage,
make up approximately 1% by volume of articular cartilage.11
The main function of chondrocytes is to produce and organize the
ECM, comprising collagen (60% of dry weight), proteoglycans
(2535% of dry weight), and non-collagenous proteins (1520%
of dry weight).11 Collagen is a fibrous molecule that increases
the tensile strength and organization of the tissue. Proteoglycans
have a protein core and numerous sulphated glycosaminoglycans
(sGAG) branches. The branches are made up of disaccharide
units; the type and number of these units determine the specific
properties of the proteoglycan.
The major types of proteoglycans are chondroitin sulfate,
keratan sulfate, dermatan sulfate, heparan sulfate, and
hyaluronan. Hyaluronan is not sulphated or attached to a protein
core, but it is the most abundant proteoglycan in cartilage and
plays a role in binding other proteoglycans in order to form
larger complexes.12 Proteoglycans have a negative charge that
attracts ions, causing an osmotic imbalance. This imbalance
leads to absorption of water which helps hydrate the tissue and
induces a swelling pressure that increases the tissue's resistance
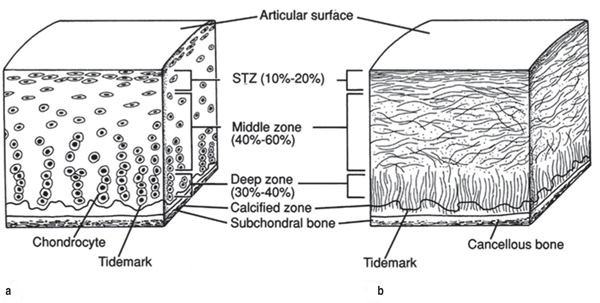
to compression.12 Cartilage also has a unique zonal organization
(Figure A). Chondrocytes and collagen fibers at the surface of the
cartilage are aligned parallel to the surface to resist shear stresses
in the superficial zone. The chondrocytes and collagen fibers are
more randomly aligned in the middle zone in order to distribute
the load throughout the tissue. Finally, the cells and fibers are
aligned perpendicular to the surface to resist compressive forces
in the deep zone.14 Clearly, although cartilage appears to be a
simple aneural, avascular, connective tissue, there are many
levels of complexity in its composition and structure.
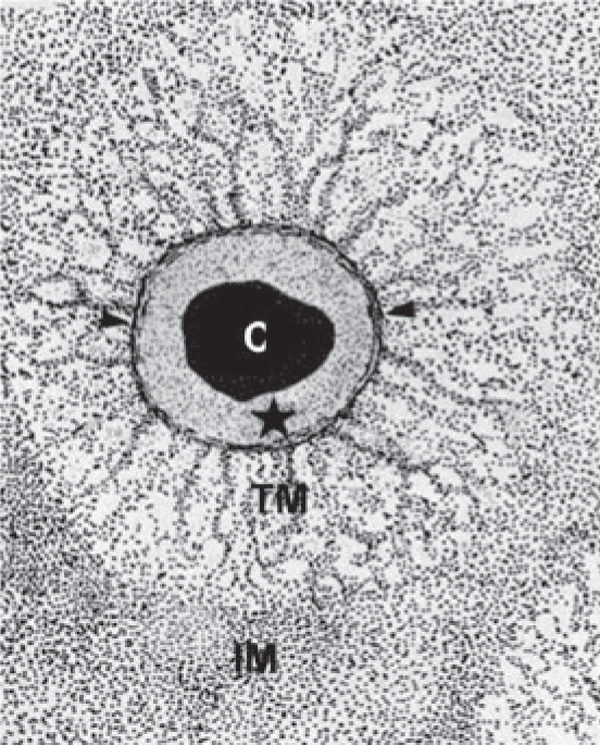
While cartilage has vertical organizational variation, differences
in matrix composition that radiate from the chondrocytes are
also important. Three regions of the ECM are distinguished by different proteins and functions: the interterritorial, territorial,
and pericellular matrices (Figure B). The interterritorial matrix
occupies the spaces furthest from the cells and consists of keratin
sulfate-rich proteoglycans and thick collagen bundles. The
territorial matrix contains chondroitin sulfate-rich proteoglycans
and smaller more radially organized collagen bundles. Finally,
the pericellular matrix (PCM) is made up of small diameter
collagen fibers that form a tightly woven capsule immediately
adjacent to the cell.15 Type II collagen is the most abundant
collagen in articular cartilage, but it is accompanied by types XI,
IX, VI, III, XII, XIV, and X collagen as well.16 Although type VI
collagen is a small percentage of the total collagen content of
articular cartilage, it has been shown to be highly concentrated
in the PCM. Type VI collagen is believed to provide pericellular
architecture and also to improve signaling between the cell and
its microenvironment.10 The PCM also consists of sulphated
proteoglycans, an assortment of glycoproteins, hyaluronan,
biglycan, fibronectin, and laminin.10 In native cartilage tissue,
cells interact with the distinct extracellular matrix in the
pericellular microenvironment. Providing an environment in
which the cells reproduce this surrounding structure may be
critical to the success of cartilage tissue engineering.
|
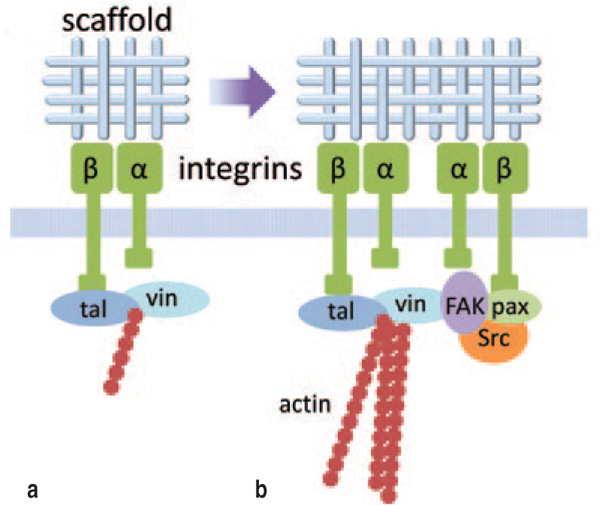
The primary linkage between the
tissue engineering scaffold and the
interior of any cell occurs through
integrin molecules on the cell surface
(Figure 1a). The integrin family
comprises cell adhesion receptors
that span the cell membrane and
bind to a wide range of extracellular
matrix (ECM) components and
scaffold materials.17 Internal to the
cell, integrins attach to the actin
cytoskeleton through intermediate
proteins.18,19 Integrins consist of two
non-covalently associated subunits,
denoted a and b. To date, 18 a and
8 b subunits have been identified, which
occur in 24 different combinations.20
The particular combination of the a
and b subunits determines the ligand
binding specificity of the integrin.
Some integrin receptors bind to a
limited number of protein sequences,
but most bind to many sequences
which could be part of the same or
different macromolecules of the ECM
or scaffold.21 Each cell type expresses a
specific subset of the known integrins,
which controls the cells' interactions
with their microenvironment.21,22
Additionally, different matrices or
scaffolds, or different arrangements of
the same matrix or scaffold component,
can transmit distinct signals to a cell
through the same integrin.21 Cellular
attachment to the ECM or scaffold
via integrin binding is essential for
migration, proliferation, differentiation,
and survival in a variety of cell types,21,2328 and as a result integrins play a
role in embryonic development; tissue
growth, remodeling and maintenance;
and in tissue engineering.
Integrin attachments modulate
cellular processes mainly through focal
adhesions (Figure 1b). Focal adhesions
are large clusters of integrins, often
located at the periphery of the cell, that
form in response to integrin binding.
In addition to assuring cell-scaffold
attachment, these integrin-based focal
adhesion complexes also provide an
intracellular concentration of more
than 50 proteins29 including scaffolding
proteins, GTPases and enzymes which
provide critical signaling between
the cell exterior and interior.30 Focal
adhesions are also a targeted location
for the anchorage of actin filaments,
a key component of the cytoskeletal
structure of most cells, which are linked
to the b integrin subunits through
various proteins.31,32 The interactions
between focal adhesions and the actin
cytoskeleton are bidirectional: actin
stress fibers regulate the assembly and
growth of the focal adhesions, while the
focal adhesions regulate the assembly
of the actin cytoskeleton.33 In some
cell types, focal adhesions have been
shown to be the site of a concentration
of growth factor receptors such as
those for basic fibroblast growth
factor, platelet-derived growth factor
and epidermal growth factor; focal
adhesion formation subsequently leads
to an enhanced response to growth
factor stimulation.34,35 Focal adhesions
also play a key role in sensing the
stiffness and surface topography of the
scaffold to which they are attached.
Therefore, scaffold stiffness and
surface topography contribute to the
lineage commitment of undifferentiated
stem cells and to the maintenance of a
differentiated morphology via focal
adhesions.33,3638
A key regulator of focal adhesion
assembly and signaling is focal
adhesion kinase (FAK). FAK is a
tyrosine kinase that localizes only
to focal adhesions39 and has been
implicated as a mechanosensor.40
Following attachment, integrin
conformation is shifted, allowing FAK
interaction with b1 integrin, resulting
in autophosphorylation of FAK at Y397
and kinase activation.41 Once activated,
FAK phosphorylates other scaffolding
proteins and triggers a signaling
cascade.42,43 FAK contains protein
binding domains and also serves as a
scaffolding protein within the focal
adhesions, independent of kinase
activity.44 Following FAK recruitment
to the focal adhesion and activation,
the resulting phosphorylation of target
proteins activates cell proliferation,
survival and migration pathways.4548
This is a fraction of FAK's potential
role in focal adhesion signaling, as
FAK activation and downstream
pathways are extremely complex.
CHONDROCYTE-MATRIX INTERACTIONS
Integrin signaling has been studied
extensively in articular chondrocytes;it has been implicated in cartilage
tissue maintenance and arthritis,49,50
and genetic mutations affecting
integrin expression result in cartilage
abnormalities and disease.51 Cell-matrix
interactions are necessary
for survival, proliferation, and
differentiation to the chondrocyte
phenotype.52 Chondrocytes have been
shown to attach to PCM proteins
through integrin binding in experiments
that use specific inhibitors of integrin-mediated
attachment. The addition
of either synthetic peptide arginine-glycine-aspartic acid (RGD) or
antibodies that block the function of b1
or b3-integrins inhibits cell attachment
to ECM proteins such as fibronectin,
vitronectin, and collagen II.53
In cartilage development,
cell attachment to fibronectin is
particularly important. Investigations
with chick embryonic limb bud cells
have determined that cell adhesion
to fibronectin via integrin binding is
necessary for condensation, which
is the first step in developmental
chondrogenesis.54,55 However, as the
cells differentiated into chondrocytes,
fibronectin expression decreased,
which implies that integrin binding may
be important in early differentiation,
but not as necessary to maintain the
differentiated state.54 Fibronectin
expression in chick limb bud cells is
modulated by members of the TGF-b
family; when TGF-b was added to these
cells prior to condensation, fibronectin
expression and chondrogenic
differentiation increased, but when
it was added after condensation the
increased fibronectin expression had no
effect on differentiation. These results
further confirm that cell-fibronectin
interactions, and therefore integrin
binding, are necessary for initial
condensation and early differentiation
but not for maintenance of the
chondrocytic phenotype.56 These results
suggest that providing the optimal
cell attachment to a scaffold may be
particularly critical when engineering
tissues from undifferentiated cells such
as mesenchymal stem cells (MSCs).
COMMON SCAFFOLDS IN CARTILAGE TISSUE ENGINEERING
A large variety of scaffolds have been used in cartilage tissue engineering, each with strengths and
weaknesses with respect to the design
criteria stated earlier. One classification
scheme for the different scaffolds is
based on whether they promote cell
attachment. For example, many of the
natural hydrogels that are common in
cartilage tissue engineering research,
such as alginate and agarose, do
not promote cell attachment. Other
materials such as collagen and fibrin
are known to promote integrin binding
and have shown promise in cartilage
repair strategies.
Non-Adherent Materials
Natural Hydrogels
Alginate and agarose are linear
polysaccharides derived from
seaweed and algae. Agarose as a
scaffold has been shown to both
promote chondrogenic induction,
and maintain the induced cartilage
phenotype.57 Agarose requires higher
than physiologic temperatures to melt,
but due to the development of low-melting
point agarose, the cell viability
concerns with the high temperatures
have been overcome.58 Alginate gels,
on the other hand, form by the addition
of divalent cations such as Sr2+, Ba2+,
and Ca2+, with calcium ions being the
most common due to their physiologic
nature and availability.59 Like many
hydrogels, there is no integrin binding
between cells and either alginate or
agarose, and encapsulated cells retain
their rounded shape. The retention of
the round cell shape and lack of cell
attachments enhances chondrogenesis
relative to cells seeded in monolayer.
Dedifferentiated cells cultured in
monolayer but covered in agarose
showed no tendencies towards a
chondrogenic phenotype, whereas
cells encapsulated in an agarose gel
differentiated into chondrocytes.
This evidence supports the idea that
agarose does not directly affect the
chondrogenic phenotype, but rather
promotes chondrogenesis based on
secondary effects such as cell shape
and attachment.60 The main weakness
of alginate and agarose hydrogels
as scaffolds for cartilage tissue
engineering is their poor mechanical
properties; they have a compressive
moduli approximately 15% of native
tissue.61 A further concern is that a
four-fold increase in the amount of
collagen type VI was observed around
chondrocytes embedded in agarose as
compared to native tissue; the collagen
fibers also ran perpendicular to the
cell surface rather than tangentially as
in native cartilage. The differences in
composition and organization could
have significant impacts on the tissue
engineering of cartilage.62
Synthetic Hydrogels
Natural hydrogels, such as alginate
and agarose, have advantages in their
inherent biocompatibility, but synthetic
and photocrosslinkable hydrogels offer
greater control of the final macroscopic
properties of the gel. Poly(ethylene
glycol) and poly(vinyl alcohol) can
be modified for photopolymerization,
which enables spatial and temporal
control of the gelation process and also
provides a mechanism to polymerize
the gel in situ (i.e., in a tissue defect).63
Photopolymerized PEG hydrogels have
been shown to support MSC survival,
differentiation, and accumulation of
chondrogenic ECM in the presence of
TGF-b1.64
Cell Adherent Materials
Collagen
Collagen is the most abundant protein
in mammals and is composed of fibrils
made up of tropocollagen triple helices.
These triple helices allow collagen to
withstand high tensile forces.12 Cells
adhere to collagen through integrin
binding, which could lead to a spread
and flattened morphology and to
chondrocyte dedifferentiation over
time. Collagen types I and II may
be fabricated into fibrous sponges
through lyophilization or hydrogels
by neutralizing an acidic collagen
solution. Collagen sponges seeded with
bovine articular chondrocytes showed
superior chondrogenesis relative to
cells seeded in monolayer based on
sGAG accumulation and chondrogenic
gene expression. The porous structure
of the collagen sponges allows cells to
aggregate and therefore they may retain
their native spherical morphology.65
Cell-seeded collagen gels contract a
large amount which can be problematic
for tissue engineering applications,
but methods have been developed to
reduce this contraction.66,67 Human
synovium-derived MSCs seeded in a
collagen hydrogel have been shown
to increase both chondrogenic gene
expression and protein synthesis in the
presence of BMP-2 and TGF-b3.68
Fibrin
Fibrin is a naturally occurring
protein involved in wound healing.
Fibrinogen, when combined with
thrombin, polymerizes into a fibrin
hydrogel. Cells attach to fibrin,
and MSCs seeded in fibrin gels
in the presence of chondrogenic
growth factors differentiate to the
chondrogenic lineage.69 Fibrin gels
may also enhance collagen synthesis
relative to type I collagen gels in
some cell types.70 One of the main
disadvantages of fibrin is that it
contracts when seeded with stem cells,
leading to poor control over the size of
the implanted scaffold. However, this
problem is diminished when fibrin is
seeded with chondrocytes.71 Fibrin
also tends to degrade quickly when
implanted, but addition of aprotinin or
e-amino-n-caproic acid slows fibrin's
rate of degradation.72,73
MODULATING CELL ATTACHEMENT VIA SCAFFOLD DESIGN
Modulating cell-scaffold interactions
through alterations of the scaffold
material allows for investigations into
the influence of integrin attachment
on cartilage matrix production. The
choice of scaffold material(s) directly
determines cell adhesion, which
has significant effects on other cell
functions, such as proliferation and
differentiation. Scaffolds composed of
natural polymers such as fibrin74,75 and
collagen7679 are often used to achieve
direct cell anchorage (Figure 2a).
These macromolecules provide good
cell adhesion, making them an obvious
scaffold material when cell attachment
is desired. Biodegradable synthetic
polymers such as poly(alpha-hydroxy
esters), poly(lactic acid, PLA),
poly(glycolic acid, PGA), and their
copolymer poly(lactic-co-glycolic
acid) (PLGA) are attractive candidates
for cartilage tissue engineering
scaffolds that promote cell attachment
in an indirect manner. These synthetic
polymers are usually hydrophobic,
allowing a protein layer to quickly
adsorb on the scaffold surface upon
contact with body fluid in vivo or
culture medium in vitro to establish cell
adhesion sites (Figure 2b).80 However,
their high hydrophobicity may impair
cell adhesion due to denaturing of the
adsorbed proteins and inaccessibility
of the adhesion motifs to cells.81 In
addition, the hydrophobicity directly
inhibits cell seeding in porous
scaffolds, because the penetration of
the aqueous cell suspension is retarded,
resulting in uneven cell distribution.82,83
Surface modifications for improved
wettability can not only effectively
promote cell seeding efficiency but also
sustained cell adhesion. Typical surface
modification methods include ethanol
treatment,84 alkaline hydrolysis,85
and plasma oxidization.86 In plasma
oxidization of polystyrene, for
example, the outermost surface of the
polymer is disrupted and hydrophilic
functional groups are produced which
can interact strongly with proteins
such as fibronectin87,88 and collagen89
to improve cell anchorage. Ultimately,
a balance between hydrophobicity
and hydrophilicity is required for cell
attachment through protein adsorption;
Groth et al.81 identified the optimal
water contact angle as 5575º based on
a fibroblast study.
Grafting ECM proteins such as
collagen to a hydrophobic surface is
another attractive technique to improve
cell seeding efficiency and promote cell
adhesion as well. A collagen coating
simultaneously produces two beneficial
effects: it increases hydrophilicity
to facilitate cell infiltration and
homogenous cell distribution, and it
directly provides cell anchorage sites on
the scaffold surface.82,89,90 For instance,
PLGA sponges were coated with a thin
collagen layer by introducing a collagen
solution under vacuum followed by
centrifugation and freeze drying.82
Collagen may also be incorporated
into PLGA scaffolds in the form of
a microsponge that fills the scaffold
pores.83 The resulting hybrid scaffold
significantly promoted cartilage matrix
production and mechanical properties
of the engineered tissue.83
Hydrogels are advantageous for
tissue engineering scaffolds because
they encapsulate cells homogenously
and provide intrinsic porosity, but many
hydrogels such as alginate, hyaluronan,
agarose and chitosan lack cell adhesion
motifs to support direct cell anchorage
(Figure 2c). Additionally, unlike rigid
polymers, the highly hydrated state
of the hydrogel molecules inhibits
adsorption of adhesive proteins from
the environment.91 The encapsulated
cells generally maintain spherical
shapes in the gels and are unable
to spread or migrate. The lack of
anchorage to the gel matrix may
adversely affect the behaviors of stem
cells such as MSCs which proliferate
only to a limited extent in the non-cell
adhesive gels.91,92 Some effort has been
made to blend non-adhesive hydrogels
with cell adhesive macromolecules
such as collagens and their denatured
form gelatin to achieve cell attachment
in hydrogels.93,94
A recent study showed that the mere
manipulation of surface topography
may change the nonadherent hydrogel
poly(ethylene glycol) to be cell
adhesive; significant cell adhesion
was induced with microscale surface
patterns, while there was no cell
adhesion to smooth substrates of the
same material.95 One explanation for
these results is that the microtopography
may have enhanced the adsorption of
adhesive proteins to the surface of the scaffolds, although the mechanism
for this is not yet known. Clearly,
material-protein interactions and the
consequent effects on cell adhesion
are complex, and more study is needed
to fully understand the optimal design
parameters for tissue engineering
scaffolds.
Synthetic peptides, particularly
those containing the arginine-glycineaspartic
acid (RGD) sequence, have
been incorporated into a variety of
biomaterials to provide natural cellmatrix
interactions that are effective at
enhancing cell adhesion and biological
activity23 (Figure 2d). The RGD
motif was identified as the minimum
essential cell adhesion peptide
sequence in fibronectin.96 Since then,
the RGD peptide sequence has been
found in ECM components such as
vitronectin, fibrinogen, von Willebrand
factor, thrombospondin, laminin,
entactin, tenascin, osteopontin,
bone sialoprotein, and under some
conditions, collagens.97 The RGD
sequence is the most effective and
most commonly used synthetic peptide
sequence for studies of cell adhesion
due to its widespread distribution
throughout the organism, its biological
impact on cell behavior and survival,
and the fact that it is a ligand for
multiple integrins.23 Cells seeded
on a non-adhesive hydrogel surface
that has been conjugated with RGD
peptides will attach and spread through
integrin binding.23,98100 Other peptide
sequences, such as the GFOGER
sequence from collagen, also promote
beneficial cell-matrix interactions.79,101
Although synthetic peptides lack the
specificity and function of native
ECM proteins, they have important
advantages when conjugated to nonadhesive
hydrogels, as they allow
direct control over ligand type and
density. Because the synthetic peptides
are typically designed to represent only
a single motif, in some cases they can
be chosen such that they selectively
activate particular adhesion receptors,
allowing for a focused investigation
of signaling pathways.23 The RGD
peptide sequence, however, is bound
by nearly half of the 24 integrins,
including α5β1, α8β1, αIIbβ3, αvβ1, αvβ3,
αvβ5, αvβ6, αvβ8, and others that show
weaker affinity.97
EFFECT OF CELL ATTACHEMENT ON CARTILAGE TISSUE ENGINEERING
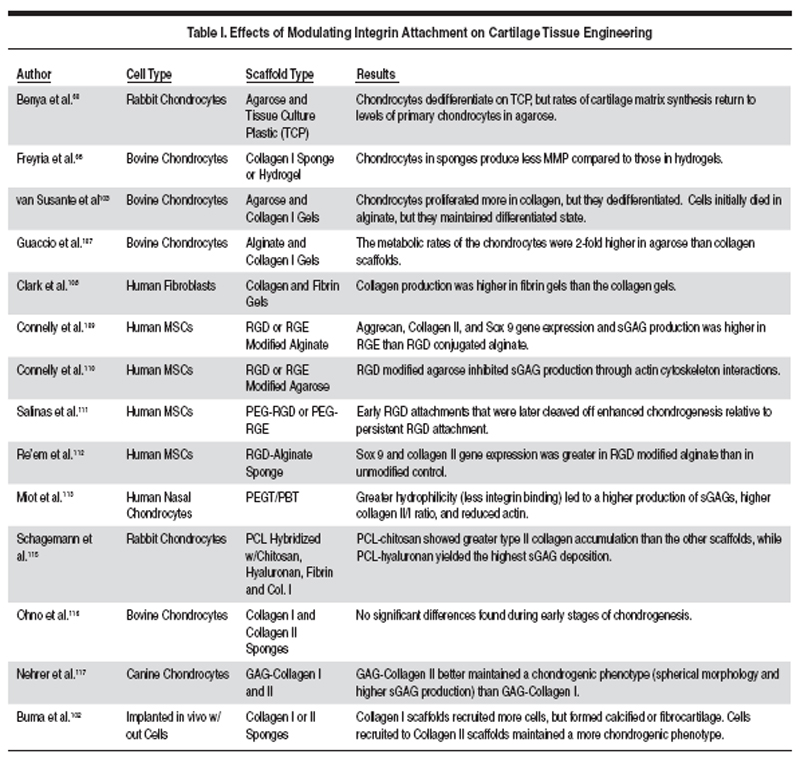
Although cartilage is one of the
few tissues for which cell attachment
to the scaffold is not absolutely
necessary for tissue engineering,
cell-scaffold interactions can both
enhance and diminish the effectiveness
of matrix production (Table I). The
role of cell attachment in cartilage
tissue engineering is complicated,
and depends on the scaffold, culture
conditions, and stage of differentiation
of the cells. For example, cell
attachment via integrin binding may
be necessary for MSCs to undergo
chondrogenesis via condensation.54,55
Additionally, scaffolds that promote
cell attachment, such as collagen gels,
enhance chondrocyte proliferation,
which could be beneficial in promoting
tissue growth.92 On the other hand,
not all effects of cell attachment
increase cartilage matrix deposition.
For example, long-term integrin
binding can lead to chondrocyte
dedifferentiation and formation of
calcified cartilage or fibrocartilage,103
while mature chondrocytes
implanted in non-adherent alginate
or agarose hydrogels maintain their
phenotype for long periods without
undergoing dedifferentiation.60,103
Furthermore, MSCs and adipose-derived
stromal cells (ASCs), which
are generally anchorage-dependent,
undergo significant chondrogenic
differentiation in alginate or agarose
hydrogels both in vitro and in vivo.104106
Cells seeded in agarose were more
metabolically active than those seeded
in collagen gels, which implies that a
lack of cellular attachment enhances
biosynthesis.107 However, cells seeded
in fibrin hydrogels increased collagen
deposition relative to those in collagen
gels as well.108 Clearly, distinguishing
the effects of cell attachment in
different materials can be difficult due
to many inherent material differences.
Therefore, the ability to modify a
material to either enhance or inhibit
cell attachment can better elucidate the
role of cell attachment as opposed to
comparing two distinct materials.
Conjugation of the integrin-attachment motif RGD to a scaffold
that otherwise does not promote cell
attachment provides a direct method
of altering integrin binding and has
been employed in several studies
investigating chondrogenesis of MSCs
for cartilage tissue engineering. MSCs
seeded in RGD modified alginate
gels expressed significantly lower
levels of the chondrogenic genes
aggrecan, collagen II, and Sox 9,
and accumulated less sGAG than
in the control samples of alginate
conjugated to non-binding RGE. These
results suggest that RGD and integrin
attachment inhibited chondrogenesis
of MSCs.109 In a follow-up study, MSC
attachment to RGD-modified agarose
gels inhibited sGAG production in a
density-dependent manner through
the RhoA/Rock pathway and actin
cytoskeleton interactions.110 In a
separate investigation, however, it was
shown that cell attachment in early
differentiation stages was beneficial
to MSC chondrogenesis. Using PEG
hydrogels conjugated to enzymatically
cleavable RGD peptides that are
released by the MSCs in later stages of
differentiation, this study demonstrated
enhanced chondrogenesis when the
MSCs bind initially but later release the
RGD attachments relative to persistent
cell attachments to uncleavable
RGD.111 Another research group
demonstrated beneficial effects of cell
attachment with lyophilized RGDalginate
sponge scaffolds for cartilage
tissue engineering. Human MSCs
seeded on the RGD sponges attained a
flattened morphology and formed focal
adhesions; the RGD group also showed
higher expression of the chondrogenic
genes Sox 9 and collagen II than
the control. They concluded that the
cells seeded in unmodified alginate
clustered together, but the cells in the
RGD-alginate attached and stayed
separated, which could have enhanced
nutrient flow to the cells and led to
a more chondrogenic phenotype.112
Cumulatively, these results using
RGD to modulate cell attachment are
somewhat confusing, as some studies
report an inhibition in chondrogenesis
with integrin binding, and others
report an enhancement. However,
the different studies used different
cell types, scaffold architectures, and peptide concentrations, which may
explain the difference in the results.
The scaffold architecture in particular
could affect the number of cell
attachment sites, nutrient availability,
cell shape, and cell aggregation which
could have a significant effect on
cellular behavior and chondrogenic
potential. Additionally, these RGD
studies demonstrate that cell-scaffold
attachment is important in cartilage
tissue engineering, but has yet to be
optimized.
Alterations to a scaffold's physiochemical
structure can also allow
insight into the role of cell attachment
on chondrogenesis. PEGT/PBT
copolymer scaffolds were constructed
and their wettability modified, while
the overall porosity and composition
of the scaffolds remained equal. The
more hydrophilic scaffolds adsorb
less protein (specifically fibronectin
which strongly promotes integrin
attachments), and the cells maintained
a more spheroidal morphology,
produced more proteoglycan, had a
higher collagen type II/I ratio, and
had reduced staining for actin relative
to the scaffold with a less hydrophilic
surface.113 Similarly, hybrid scaffolds
composed of polycaprolactone and
biopolymers such as hyaluronan,
chitosan, fibronectin, and collagen
were fabricated in order to explore the
effect of cell attachment with specific
ECM molecules.114,115 These scaffolds
demonstrated that chondrocyte
interaction with hyaluronan and chitosan
promotes neocartilage formation and
may be promising components of a
cartilage tissue engineering scaffold.114
Therefore, by modifying the protein
adsorption or bioactivity of a scaffold,
insights into the role of cell attachment
in chondrogenesis may be better
elucidated.
Although type I and II collagen both
support cell attachment, differences
in the chemical structures may lead to
cell binding through different integrins
or to different spatial arrangements of
integrin attachments when cells are
seeded in fibrous collagen sponges and hydrogels. Although type II
collagen is more abundant in native
cartilage than type I, no significant
differences were found between type
I and type II collagen sponges seeded
with chondrocytes after 20 days of
in vitro culture.116 GAG-collagen
copolymer sponges were formed
using either collagen type I or II, and
the GAG-collagen type II sponge
maintained a chondrogenic phenotype
better than the GAG-collagen type
I copolymer.117 Both collagen type I
and II sponges were implanted into
full thickness defects in rabbit knees.
Collagen type I sponges were found
to recruit more subchondral progenitor
cells than collagen II sponges, but
promoted calcified cartilage and
fibrocartilage formation. Collagen type
II sponges, however, supported a more
chondrogenic phenotype.102 Therefore
a matrix composed of both cell types
with a deep layer of type I collagen and
a superficial layer of type II collagen
was proposed.
Inhibition of the signaling molecules
that are activated by cell attachment,
such as focal adhesion kinase (FAK),
can also add insight into the effects
of cell attachment on chondrogenesis.
While the addition of exogenous type
II collagen to chondrocytes increased
FAK signaling, knockdown of FAK
by siRNA inhibited expression of
type II collagen and cell proliferation,
indicating that FAK is required for
communication with collagen II
and is involved in the regulation of
type II collagen expression and cell
proliferation.118 However, suppression
of FAK also enhances the early stages
of chondrogenesis; FAK inhibition
in micromass cultures increased
expression of chondrogenic genes,
suggesting that cell-cell interactions
rather than cell-ECM interactions
drive condensation and early
chondrogenesis.119
CONCLUSIONS
Although current cartilage tissue
engineering strategies use a wide
range of scaffold materials, the above
data demonstrate that cell adhesion is
involved in the regulation of cartilage
matrix production in engineered
tissues. Providing the correct
microenvironmental attachment signals to the cell via the scaffold
may be necessary to replicate the
complex structure and organization of
cartilage tissue. The optimal scaffold
type and surface treatment have yet
to be determined, but may vary with
cell type, stage of differentiation,
culture condition and scaffold material.
Cell type may have an especially
large influence on the effect of cell
attachment, as chondrocytes form a
PCM and quickly bind to the proteins
found there; the question of cell
attachment may be less crucial to these
cells as long as dedifferentiation is not
induced. Formation of the PCM and
its interaction with the chondrocytes
may also explain the relative success
in engineering cartilage with so many
different scaffold materials.
Although chondrocytes are an
obvious choice as a source of cells
for cartilage tissue engineering, their
limited number, slow proliferation,
as well as donor site morbidity have
led to intense interest in alternate
cell types that can differentiate to the
chondrogenic lineage, such as MSCs
and ASCs. With these undifferentiated
cells, cell-scaffold attachments may
be especially important as they may
be involved in driving the cell to the
chondrogenic differentiation pathway.
However, cell adhesion density and
type have not been optimized for
these cells, and in fact results from
the literature are in conflict on the
question of whether integrin binding is
beneficial for or inhibitory to cartilage
tissue engineering (Table I). We expect
that further, more detailed studies will
elucidate the role of cell attachment
in chondrogenesis and will enhance
efforts to engineer cell-based cartilage
therapies.
ACKNOWLEDGEMENTS
The authors acknowledge support
from the University of Notre Dame
Adult Stem Cell Initiative, the U.S.
Army Medical Research and Materiel
Command W81XWH-07-0662
and W81XWH-09-1-0741, and the
Naughton Graduate Fellowship.
REFERENCES
1. R.C. Lawrence et al., Arthritis Rheum,, 41 (5) (1998), pp. 778799.
2. J.A. Buckwalter, C. Saltzman, and T. Brown, Clin.
Orthop. Relat. Res. (427 Suppl) (2004), pp. S6S15.
3. Arthritis Foundation, Association of State and
Territorial Health Officers (1999).
4. H.J. Mankin, N. Engl. J. Med., 291 (24) (1974), pp.
12851292.
5. F. Shapiro, S. Koide, and M.J. Glimcher, J. Bone Joint
Surg. Am., 75 (4) (1993), pp. 532553.
6. S.N. Redman, S.F. Oldfield. and C.W. Archer, Eur.
Cell. Mater., 9 (2005), pp. 2332.
7. R. Langer and J.P. Vacanti, Science, 260 (5110)
(1993), pp. 920926.
8. D.L. Butler, S.A. Goldstein, and F. Guilak, J. Biomech.
Eng., 122 (2000), pp. 570575.
9. D.W. Hutmacher, Biomaterials, 21 (24) (2000), pp.
25292543.
10. C.A. Poole, J. Anatomy, 191 (1) (1997), pp. 113.
11. J.A. Buckwalter and H.J. Mankin, J. Bone and Joint
Surg., 79 (4) (1997), pp. 600611.
12. E.M. Culav, C.H. Clark, and M.J. Merrilees, Phys.
Ther., 79 (3) (1999), pp. 308319.
13. J.A. Buckwalter, V.C. Mow, and A. Ratcliffe, J. Am.
Acad. Orthop. Surg., 2 (4) (1994), pp. 192201.
14. T.J. Klein, J. Malda, R.L. Sah, and D.W. Hutmacher,
Tissue Eng. Part B: Rev., 15 (2) (2009), pp. 143157.
15. C. Poole, M.H. Flint, and B.W. Beaumont, J. Anat., 138 (Pt 1) (1984), pp. 113138.
16. D.R. Eyre, Arthritis Research, 4 (1) (2002), pp.
3035.
17. R.O. Hynes, Cell, 69 (1) (1992), pp. 1125.
18. C. Brakebusch and R. Fässler, EMBO J., 22 (10)
(2003), pp. 23242333.
19. I. Delon and N.H. Brown, Curr. Opin. Cell Biol., 19
(1) (2007), pp. 4350.
20. A. van der Flier and A. Sonnenberg, Cell Tissue
Res., 305 (3) (2001), pp. 285298.
21. D.G. Stupack and D.A. Cheresh, J. Cell. Sci., 115
(19) (2002), pp. 37293738.
22. B.H. Luo, C.V. Carman, and T.A. Springer, Annu.
Rev. Immunol., 25 (2007), pp. 619647.
23. U. Hersel, C. Dahmen, and H. Kessler, Biomaterials,
24 (24) (2003), pp. 43854415.
24. J.A. Eble and J. Haier, Current Cancer Drug
Targets, 6 (2) (2006), pp. 89105.
25. D. Docheva, C. Popov, W. Mutschler, and M.
Schieker, J. Cell. Mol. Med., 11 (1) (2007), pp. 2138.
26. K. Raymond, M.A. Deugnier, M.M. Faraldo, and
M.A. Glukhova, Curr. Opin. Cell Biol., 21 (5) (2009),
pp. 623629.
27. E.A. Clark and J.S. Brugge, Science, 268 (5208)
(1995), pp. 233239.
28. A. Howe, A.E. Aplin, S.K. Alahari, and R.L. Juliano,
Curr. Opin. Cell Biol., 10 (2) (1998), pp. 220231.
29. E. Zamir and B. Geiger, J. Cell. Sci., 114 (20)
(2001), pp. 35833590.
30. R.O. Hynes, Cell, 110 (6) (2002), pp. 673687.
31. R. Zaidel-Bar, S. Itzkovitz, A. Ma'ayan, R. Iyengar,
and B. Geiger, Nat. Cell Biol., 9 (8) (2007), pp. 858
867.
32. B. Geiger, A. Bershadsky, R. Pankov, and K.M.
Yamada, Nat. Rev. Mol. Cell Biol., 2 (11) (2001), pp.
793805.
33. B. Geiger, J.P. Spatz, and A.D. Bershadsky, Nat.
Rev. Mol. Cell Bio., 10 (1) (2009), pp. 2133.
34. G.E. Plopper, H.P. McNamee, L.E. Dike, K.
Bojanowski, and D.E. Ingber, Mol. Biol. Cell, 6 (10)
(1995), pp. 13491365.
35. S. Miyamoto, H. Teramoto, J.S. Gutkind, and K.M.
Yamada, J. Cell Biol., 135 (6) (1996), pp. 16331642.
36. A.J. Engler, S. Sen, H.L. Sweeney, and D.E.
Discher, Cell, 126 (4) (2006), pp. 677689.
37. D.E. Discher, P. Janmey, and Y. Wang, Science, 310
(5751) (2005), pp. 11391143.
38. V. Vogel and M. Sheetz, Nat. Rev. Mol. Cell Bio., 7
(4) (2006), pp. 265275.
39. M. Schaller, C.A. Borgman, B.S. Cobb, R.R. Vines,
A.B. Reynolds, and J.T. Parsons, Proc. Nat. Acad. of
Sci., 89 (11) (1992), pp. 51925196.
40. H.B. Wang, M. Dembo, S.K. Hanks, and Y. Wang,
Proc. Nat. Acad. of Sci., 98 (20) (2001), pp. 1129511300.
41. J. Zhao and J.L. Guan, Cancer Metastasis Rev., 28
(1) (2009), pp. 3549.
42. J.T. Parsons, J. Cell. Sci., 116 (8) (2003), pp.
14091416.
43. S. Roy, P.J. Ruest, and S.K. Hanks, J. Cell.
Biochem., 84 (2) (2002), pp. 377388.
44. M.C. Beckerle, M.D. Schaller, J.D. Hildebrand,
and J.T. Parsons, Mol. Biol. Cell, 10 (10) (1999), pp.
34893505.
45. A.P. Gilmore, T.W. Owens, F.M. Foster, and J.
Lindsay, Curr. Opin. Cell Biol., 21 (5) (2009), pp.
654661.
46. A. Gilmore and L.H. Romer, Mol. Biol. Cell, 7 (8)
(1996), pp. 12091224.
47. D. Ilic et al., Nature (London), 377 (6549) (1995),
pp. 539544.
48. L.A. Cary, J.F. Chang, and J.L. Guan, J. Cell. Sci.,
109 ( Pt 7) (1996), pp. 17871794.
49. R.F. Loeser, Biorheology, 37 (1) (2000), pp. 109
116.
50. S. Millward-Sadler and D.M. Salter, Ann. Biomed.
Eng., 32 (3) (2004), pp. 435446.
51. A. Woods, G. Wang, and F. Beier, J. Biol. Chem.,
280 (12) (2005), pp. 1162611634.
52. M.S. Hirsch, L.E. Lunsford, V. Trinkaus-Randall,
and K.K.H. Svoboda, Developmental Dynamics, 210
(3) (1997), pp. 249263.
53. R.F. Loeser, Arthritis & Rheumatism, 36 (8) (1993),
pp. 11031110.
54. S. Tavella et al., J. Cell. Sci., 110 ( Pt 18) (1997),
pp. 22612270.
55. A.L. Gehris, E. Stringa, J. Spina, M.E. Desmond,
R.S. Tuan, and V.D. Bennett, Dev. Biol., 190 (2) (1997),
pp. 191205.
56. E.F. Roark and K. Greer, Am .J. Anat., 200 (2)
(1994), pp. 103116.
57. A.Y. Thompson, K.A. Piez, and S.M. Seyedin, Exp.
Cell Res., 157 (2) (1985), pp. 483494.
58. J. Raghunath, J. Rollo, K.M. Sales, P.E. Butler, and
A.M. Seifalian, Biotechnol. Appl. Biochem., 46 (Pt 2)
(2007), pp. 7384.
59. O. Smidsrød, Faraday Discuss. Chem. Soc., 57
(1974), pp. 263274.
60. P.D. Benya and J. D. Shaffer, Cell, 30 (1) (1982),
pp. 215224.
61. J.L. Drury and D.J. Mooney, Biomaterials, 24 (24)
(2003), pp. 43374351.
62. M. Dimicco, J.D. Kisiday, H. Gong, and A.J.
Grodzinsky, Osteoarthritis and Cartilage/OARS,
Osteoarthritis Research Society, 15 (10) (2007), pp.
12071216.
63. S.J. Bryant and K.S. Anseth, J. Biomed. Mater.
Res., 59 (1) (2002), pp. 6372.
64. C.G. Williams, T.K. Kim, A. Taboas, A. Malik, P.
Manson, and J. Elisseeff, Tissue Eng., 9 (4) (2003),
pp. 679688.
65. K.E. Yates, F. Allemann, and J. Glowacki, Cell
Tissue Banking, 6 (1) (2005), pp. 4554.
66. A.M. Freyria et al., Tissue Eng. Part A, 15 (6)
(2008), pp. 12331245.
67. E. Gentleman, E.A. Nauman, K.C. Dee, and G.A.
Livesay, Tissue Eng., 10 (3-4) (2004), pp. 421427.
68. A. Yokoyama, I. Sekiya, K. Miyazaki, S. Ichinose, Y.
Hata, and T. Muneta, Cell Tissue Res., 322 (2) (2005),
pp. 289298.
69. G.I. Im, Current Applied Physics, 5 (5) (2005), pp.
438443.
70. E. Grassl, T.R. Oegema, and R.T. Tranquillo, J.
Biomed. Mater. Res. Part A, 60 (4) (2002), pp. 607
612.
71. G.M. Peretti, M.A. Randolph, M.T. Villa, M.S.
Buragas, and M.J. Yaremchuk, Tissue Eng., 6 (5)
(2000), pp. 567576.
72. Q. Ye et al., European J. Cardio-Thoracic Surg., 17
(5) (2000), pp. 587591.
73. A. Mol et al., Biomaterials, 26 (16) (2005), pp.
31133121.
74. G.M. Peretti et al., Ann. Plast. Surg., 46 (5) (2001),
pp. 533537.
75. D.A. Hendrickson et al., J. Orthop. Res., 12 (4)
(1994), pp. 485497.
76. S.R. Frenkel, B. Toolan, D. Menche, M.I. Pitman,
and J.M. Pachence, J. Bone Joint Surg. Br., 79 (5)
(1997), pp. 831836.
77. M.C. Ronziere, S. Roche, J. Gouttenoire,
O. Demarteau, D. Herbage, and A.M. Freyria,
Biomaterials, 24 (5) (2003), pp. 851861.
78. A.E. Sams, R.R. Minor, J.A. Wootton, H.
Mohammed, and A.J. Nixon, Osteoarthritis Cartilage,
3 (1) (1995), pp. 6170.
79. S.Q. Liu, Q. Tian, J.L. Hedrick, J.H. Po Hui, P.L.
Ee, and Y.Y. Yang, Biomaterials, 31 (28) (2010), pp.
72987307.
80. R.A. Latour, Encyc. Biomater. and Biomed. Eng.,
(2005), pp. 115.
81. T. Groth, G. Altankov, A. Kostadinova, N. Krasteva,
W. Albrecht, and D. Paul, J. Biomed. Mater. Res., 44 (3)
(1999), pp. 341351.
82. G. Chen et al., J. Biomed. Mater. Res. BAppl.
Biomater., 90 (2) (2009), pp. 864872.
83. W. Dai, N. Kawazoe, X. Lin, J. Dong, and G. Chen,
Biomaterials, 31 (8) (2010), pp. 21412152.
84. A.G. Mikos, M.D. Lyman, L.E. Freed, and R. Langer,
Biomaterials, 15 (1) (1994), pp. 5558.
85. J. Gao, L. Niklason, and R. Langer, J. Biomed.
Mater. Res., 42 (3) (1998), pp. 417424.
86. J. Yang, Y. Wan, J. Yang, J. Bei, and S. Wang, J.
Biomed. Mater. Res. A., 67 (4) (2003), pp. 11391147.
87. T.G. van Kooten, H.T. Spijker, and H.J. Busscher,
Biomaterials, 25 (10) (2004), pp. 17351747.
88. A.P. Kowalczyk and P.J. McKeown-Longo, J. Cell.
Physiol., 152 (1) (1992), pp. 126134.
89. J. Yang, J. Bei, and S. Wang, Biomaterials, 23 (12)
(2002), pp. 26072614.
90. Z. Ma, C. Gao, Y. Gong, and J. Shen, Biomaterials,
26 (11) (2005), pp. 12531259.
91. J.T. Oliveira and R.L. Reis, J. Tissue Eng. Regen.
Med., (2010), in press.
92. H. Yamaoka et al., J. Biomed. Mater. Res. A, 78 (1)
(2006), pp. 111.
93. D. Bosnakovski, M. Mizuno, G. Kim, S. Takagi, M.
Okumura, and T. Fujinaga, Biotechnol. Bioeng., 93 (6)
(2006), pp. 11521163.
94. C. Yang, H. Frei, F.M. Rossi, and H.M. Burt, J.
Tissue Eng. Regen. Med., 3 (8) (2009), pp. 601614.
95. V.A. Schulte, M. Diez, M. Moller, and M.C. Lensen,
Biomacromolecules, 10 (10) (2009), pp. 27952801.
96. M.D. Pierschbacher and E. Ruoslahti, Nature, 309
(5963) (1984), pp. 3033.
97. E. Ruoslahti, Ann. Rev. Cell and Devel. Bio., 12 (1)
(1996), pp. 697715.
98. B. Jeschke et al., Biomaterials, 23 (16) (2002), pp.
34553463.
99. J.A. Rowley and D.J. Mooney, J. Biomed. Mater.
Res., 60 (2) (2002), pp. 217223.
100. J.A. Rowley, G. Madlambayan, and D.J. Mooney,
Biomaterials, 20 (1) (1999), pp. 4553.
101. C.D. Reyes and A.J. García, J. Biomed. Mater.
Res. Part A, 65 (4) (2003), pp. 511523.
102. P. Buma et al., Biomaterials, 24 (19) (2003), pp.
32553263.
103. J.L. van Susante et al., Acta Orthop. Scand., 66
(6) (1995), pp. 549556.
104. C.T. Buckley et al., J. Biomech., 43 (5) (2010), pp.
920926.
105. H.L. Ma, S.C. Hung, S.Y. Lin, Y.L. Chen, and
W.H. Lo, J. Biomed. Mater. Res. A, 64 (2) (2003), pp.
273281.
106. K.H. Bouhadir, K.Y. Lee, E. Alsberg, K.L. Damm,
K.W. Anderson, and D.J. Mooney, Biotechnol. Prog., 17
(5) (2001), pp. 945950.
107. A. Guaccio, C. Borselli, O. Oliviero, and P.A. Netti,
Biomaterials, 29 (10) (2008), pp. 14841493.
108. R.A. Clark, L.D. Nielsen, M.P. Welch, and J.M.
McPherson, J. Cell. Sci., 108 ( Pt 3) (Pt 3) (1995), pp.
12511261.
109. J.T. Connelly, A.J. García, and M.E. Levenston,
Biomaterials, 28 (6) (2007), pp. 10711083.
110. J.T. Connelly, A.J. Garcia, and M.E. Levenston, J.
Cell. Physiol., 217 (1) (2008), pp. 145154.
111. C.N. Salinas and K.S. Anseth, Biomaterials, 29
(15) (2008), pp. 23702377.
112. T. Re'em, O. Tsur-Gang, and S. Cohen,
Biomaterials, 31 (26) (2010), pp. 67466755.
113. S. Miot et al., Biomaterials, 26 (15) (2005), pp.
24792489.
114. J. Schagemann et al., J. Biomed. Mater. Res. Part
A, 93 (2) (2010), pp. 454463.
115. J.C. Schagemann et al., Biomaterials, 31 (10)
(2010), pp. 27982805.
116. T. Ohno, K. Tanisaka, Y. Hiraoka, T. Ushida, T.
Tamaki, and T. Tateishi, Mater. Sci. and Eng.: C, 24 (3)
(2004), pp. 407411.
117. S. Nehrer et al., J. Biomed. Mater. Res. Part B:
Appl. Biomater., 38 (2) (1997), pp. 95104.
118. Y.H. Kim and J.W. Lee, J. Cell. Physiol., 218 (3)
(2009), pp. 623630.
119. D. Pala et al., J. Biol. Chem., 283 (14) (2008), pp.
92399247.
Andrew J. Steward, graduate student, Yongxing
Liu, visiting research assistant professor, and
Diane R. Wagner, assistant professor, are with
the Department of Mechanical Engineering and
Bioengineering Graduate Program, University of
Notre Dame, Notre Dame, IN 46556 USA. Steward
is also with the Department of Mechanical and
Manufacturing Engineering and Trinity Centre
for Bioengineering, Trinity College Dublin,
Dublin, Ireland. Dr. Wagner can be reached at
the Department of Aerospace and Mechanical
Engineering, 145 Multidisciplinary Research
Building, Notre Dame, IN 46556 USA; (574) 631-
5735; e-mail: dwagner@nd.edu. |
 Presenting a Web-Enhanced
Presenting a Web-Enhanced