
| Next Session |
|---|
| Return To Program Contents Page |
Room: 330G
Session Chairperson: Elizabeth A. Holm, Sandia National Lab, Physical and Joining Metallurgy, MS 1411, Albuquerque, NM 87185-0340
8:30 am INVITED
THEORETICAL CALCULATIONS FOR INTERFACES: GRAIN BOUNDARIES IN COVALENT MATERIALS AND METAL-CERAMIC INTERFACES: Masanori Kohyama, Dept. of Material Physics, Osaka National Research Institute, AIST, 1-8-31, Midorigaoka, Ikeda, Osaka, 563, Japan
For distorted configurations at interfaces in covalent materials or for interfaces between dissimilar materials, it is difficult to develop reliable interatomic potentials. For such systems, it is desirable to perform electronic structure calculations so as to clarify the stable configurations and microscopic nature. Currently, such kinds of enormous calculations are becoming possible by virtue of the development of the efficient theoretical methods and the high-performance computers. In this paper, we present our recent band-theoretical calculations for grain boundaries in Si, SiC and diamond, and for Al-SiC interfaces. First, grain boundaries in covalent materials have been dealt with by using the transferable tight-binding method [1]. For those in Si, general features of stable configurations and general relations between local structural disorder and local electronic structure are clarified. These are compared with those in diamond. For those in SiC, effects of interfacial C-C or Si-Si bonds and interfacial stoichiometry are analyzed. Second, ab initio calculations for grain boundaries in Si and in SiC have been performed by using the first-principles molecular dynamics (FPMD) method [2]. Stability of the structural models and the nature of interfacial C-C and Si-Si bonds in SiC are analyzed more quantitatively. Third, ab initio calculations for Al-SiC interfaces have been investigated [3]. The features of Al-C and Al-Si interactions at the interfaces are clarified.
9:10 am
ATOMIC STRUCTURE OF INTERFACES IN THE LAMELLAR TiAl AND EFFECTS OF DIRECTIONAL BONDING: V. Vitek, R. Siegl, Department of Materials Science and Engineering, University of Pennsylvania, Philadelphia, PA 19104; H. Inui, M. Yamaguchi, Department of Metal Science and Technology, Kyoto University, Kyoto 606, Japan
The microstructure of nearly stoichiometric TiAl alloys, which are important candidates for high-temperature applications, consists of lamellae with L10 and DO19 structures. A serious limitation is their low room temperature ductility. The failure often occurs by cracking along the lamellar interfaces and one clue to the fracture propensity is the atomic structure of these interfaces. To investigate this atomic structure we first carried out atomistic calculations employing Finnis-Sinclair type central force many-body potentials. However, calculated atomic structures exhibit some significant discrepancies when compared with high-resolution electron microscopy observations. For this reason further calculations have been made using an ab initio electronic structure full-potential method. An excellent agreement between calculated and observed structures was then attained. This result can be interpreted in terms of the covalent type bonding across the interface. This emphasizes significance of directional bonding in TiAl which may play an important role in its mechanical behavior. *Research supported by the U.S. Dept. of Energy, Office of Basic Energy Sciences, Grant No. DE-FG02-87ER45295 and NEDO.
9:30 am
ATOMISTIC STUDY OF GRAIN BOUNDARIES IN NiAl: M. Yan, V. Vitek, S.P. Chen, Theoretical Division, Los Alamos National Laboratory, Los Alamos, NM 87545; Department of Materials Science and Engineering, University of Pennsylvania, Philadelphia, PA 19104
We have studied the grain boundary properties in NiAl B2 compound by applying the empirical N-body central force potentials of the Finnis-Sinclair type. These potentials have been constructed for B2 NiAl by fitting a number of equilibrium properties of the alloy and reproducing the asymmetric behavior of constitutionals point defects in off-stoichiometric NiAl. At the same time, these potentials assure the structural and mechanical stability of the B2 lattice. It was found that in stoichiometric NiAl alloy boundaries in surplus of Al atoms have appreciably lower cohesive strength than the stoichiometric boundaries or boundaries in surplus of Ni atoms. From the structural point of view, boundaries in surplus of Al possess the largest expansion and are associated with large "holes". On the other hand, boundaries with the stoichiometric configuration or in surplus of Ni atoms have more compact structures. The segregation of the antisite defects and vacancies at grain boundaries was also investigated by performing molecular static and Monte Carlo calculations. It was found that no antisite defects are favored at grain boundaries at the stoichiometric bulk composition. In the case of Ni in surplus in the bulk Al atoms have strong tendency to segregate to the boundary region. The effects of such grain boundary features upon the mechanical properties of NiAl will be discussed.
9:50 am
GRAIN BOUNDARY STRUCTURE TRENDS IN B2 COMPOUNDS: Batsirai Mutasa, Diana Farkas, Yuri Mishin, Department of Materials Science and Engineering, Virginia Polytechnic Institute and State University, Blacksburg, VA 24061
The relaxed atomistic grain boundary structures in B2 aluminides were investigated using molecular statics and embedded atom potentials in order to explore general trends for a series of B2 compounds. Free surface energies and grain boundary structures were studied in three compounds, FeAl, NiAl and CoAl. These alloys respectively have increasing anti-phase boundary energies. The misorientations chosen for detailed study correspond to the 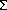 5(310) and 5(210) boundaries. The effects of both boundary stoichiometry and simulation block stoichiometry on grain boundary energetics were considered. Chemical potentials and point defect energies were calculated for boundaries contained in both stoichiometric and off-stoichiometric bulks. The surface energies for these B2 aluminides were also calculated so that trends concerning the cohesive energy of the boundaries could be studied. The implications of stoiciometry, the multiplicity of the boundary structures and possible transformations between them for grain boundary brittleness are also discussed.
5(310) and 5(210) boundaries. The effects of both boundary stoichiometry and simulation block stoichiometry on grain boundary energetics were considered. Chemical potentials and point defect energies were calculated for boundaries contained in both stoichiometric and off-stoichiometric bulks. The surface energies for these B2 aluminides were also calculated so that trends concerning the cohesive energy of the boundaries could be studied. The implications of stoiciometry, the multiplicity of the boundary structures and possible transformations between them for grain boundary brittleness are also discussed.
10:10 am BREAK
10:30 am INVITED
EPITAXY AND ORIENTATION RELATIONSHIP IN BICRYSTALS: P. Pirouz, Y. Ikuhara, F. Ernst, Deparment of Materials Science and Engineering, Case Western Reserve University, Cleveland, OH 44106-7204; Department of Materials, The University of Tokyo, Bunkyo-ku, Tokyo 113, Japan; Max-Planck-Institut für Metallforschung, Seestr. 92, D-70174, Stuttgart, Germany
When two crystals are in contact, the interaction between the atoms at or near their interface often orients the crystals in a unique way. Usually, one of the two crystals is a matrix or a substrate, and the other crystal is a precipitate within the matrix, or a deposit on the substrate. In this talk, a recently-proposed method for predicting the orientation relationship (OR) between two crystals is described and experimental cases of film/substrate composites are compared with the predictions of the model. The case of three-dimensional OR, where the film-substrate orientation relationship is maintained irrespective of the substrate surface on which the film is grown, is described, and this is compared with those cases where multiple ORs may be favored resulting on the dependence of the OR on the substrate surface. Finally, the physical basis of the model is described in terms of the energy minimization of adatoms deposited on a rigid substrate.
11:10 am
STRUCTURAL CHARACTERIZATION AND MODELING OF THE ALUMINUM {111}/SAPPHIRE (0001) HETEROPHASE INTERFACE: D.L. Medlin, K.F. McCarty, R.Q. Hwang, J.E. Smugeresky, T. Tsuji and M.I. Baskes, Sandia National Laboratories, Livermore CA 94551; Shizuoka University, Jyoohoku 3-5-1, Hamamatsu 432, Japan
We are investigating Al/Al2O3 interface structure using thin films fabricated by deposition of aluminum from an effusion source onto (0001) sapphire under UHV conditions. Structural considerations suggest that the metal would grow to match the close-packed metal planes and directions with the close-packed oxygen ions of the sapphire substrate, i.e., (0001)Al2O3//(111) metal and [10-10]Al2O3//[-110]metal. Although this orientation is predominant, transmission electron microscopy observations also show the existence of two additional types of orientation relationship, corresponding to rotations of 30 degrees and ~11 degrees from the primary domain orientation. A comparison of the predictions of atomistic calculations for the interface structure and dislocation configuration with the results of high resolution and conventional transmission electron microscopy observations will be presented. This work is supported by the U.S. DOE under contract DE-AC04-94AL85000.
11:30 am
INVESTIGATION OF THE BONDING CHANGES ASSOCIATED WITH GRAIN BOUNDARY EMBRITTLEMENT: V.J. Keast, J. Bruley, D.B. Williams, P. Rez*, Department of Materials Science and Engineering, Whitaker Lab #5, Lehigh University, Bethlehem PA 18015; *Center for Solid State Science and Department of Physics, Arizona State University, Tempe, AZ 85287-1704
Grain boundary embrittlement by impurity or alloying elements is a common and technologically important phenomenon, of which the embrittlement of Cu by Bi is a classic example. Investigations, using the near edge fine structure in the electron energy loss spectrum (EELS), have indicated that there is a change in bonding at the grain boundaries associated with the segregation of Bi. Namely, there is a decrease in the density of the d states for Cu atoms at the boundary when Bi is present. The effect of variations in grain boundary misorientation on the Bi segregation levels and on the near edge structure will be described. Comparisons between results from EELS and x-ray photoelectron spectroscopy (XPS) will also be made. (Supported by NSF) (DMR 93-0625).
| Next Session | ||||
|---|---|---|---|---|
| Search | Technical Program Contents | 1997 Annual Meeting Page | TMS Meetings Page | TMS OnLine |