
![]() Focusing on physical metallurgy and materials, Materials Week '97, which incorporates the TMS Fall Meeting, features a wide array of technical symposia sponsored by The Minerals, Metals & Materials Society (TMS) and ASM International. The meeting will be held September 14-18 in Indianapolis, Indiana. The following session will be held Monday morning, September 15.
Focusing on physical metallurgy and materials, Materials Week '97, which incorporates the TMS Fall Meeting, features a wide array of technical symposia sponsored by The Minerals, Metals & Materials Society (TMS) and ASM International. The meeting will be held September 14-18 in Indianapolis, Indiana. The following session will be held Monday morning, September 15.
Program Organizers: A. Gonis, P.E.A. Turchi, Chemistry and Materials Science Department (L-268), Lawrence Livermore National Laboratory, P.O. Box 808, Livermore, CA 94551; G.M. Stocks, Metals and Ceramics Division, MS 6114, Oak Ridge National Laboratory, Oak Ridge, TN 37831
Room: 202
Session Chair: Prof. J.S. Faulkner, Alloy Research Center and Department of Physics, Florida Atlantic University, Boca Raton, FL 33431
SOME PROPERTIES OF Cu-Au ALLOY SURFACES: R.G. Jordan, Alloy Research Center and Department of Physics, Florida Atlantic University, Boca Raton, FL 33431; G.Y. Guo, CCLRC Daresbury Laboratory, Warrington WA4 4AD, UK
Experimental and theoretical studies of the low index surfaces of Cu-Au (ordered) alloys reveal a wealth of interesting details. In this talk we highlight some of our more recent results and discuss their implications with particular reference to the electronic structure of the alloys in the surface region. We focus in particular on surface states and the dependence of surface core level shifts on orientation and segregation. Supported by the NSF (#DMR-9500654) and NATO (#CRG.910981).
9:10 am INVITED
TIGHT-BINDING AND CVM CALCULATION OF PROFILES OF APB ENERGIES IN Ni3Al: T. Shinoda, Y. Mishima, P. & I. Laboratory, Tokyo Institute of Technology, Nagatsuta 4259, Midori-ku, Yokohama 227, Japan; K. Masuda-Jindo, Department of Materials Science and Engineering, Tokyo Institute of Technology, Nagatsuta 4259, Midori-ku, Yokohama 227, Japan
A tight-binding (TB) recursion scheme, coupled to LMTO method, is used to calculate the APB energy of Ni3Al and NiAl alloys. The total energy of the system is given by a sum of band structure energy and ionic repulsion. Since the electrostatic contributions cancel out, what is needed to calculate the effective pair interaction (EPI) energies is an average over all configurations, with fixed occupancy of sites i and j, of the one-electron band-structure term. The electronic structures of the alloys are calculated by tridiagonalizing a TB Hamiltonian using the continued fraction technique. We estimate the effective pair (cluster) interaction energies (EPI) using the direct configurational averaging (DCA) method [1] over a small number of randomly generated configurations. The present calculation of equilibrium profiles of (111) APB energy in Ni3Al alloys is based on the CVM with the tetrahedron cluster approximation, using the empirical pair interaction energies as well as those estimated by a tight-binding recursion scheme coupled to a LMTO method. Calculated APB energies, that is the sums of each overall profile are compared with the experimentally obtained values in their magnitudes. Some arguments are tried with respect to the superiority to each other of the two pair interaction energies.
9:50 am
PHOTOEMISSION FROM THE (111) SURFACES OF Cu-Au ALLOYS: L.R. Masliah, R.G. Jordan, S.L. Qiu, Alloy Research Center and Department of Physics, Florida Atlantic University, Boca Raton, FL 33431; B. Ginatempo, Dipartimento di Fisica and Unità di Ficerca INFM di Messina, Università di Messina, Salita Sperone 31, 98166 Messina, Italy
We have calculated the electronic structure at the (111) surfaces of ordered Cu-Au alloys using the LMTO method in a supercell geometry and including the surface dipole terms. In order to make quantitative comparisons with our photoemission measurements we have used the potential functions in relativistic photocurrent calculations. In this talk we concentrate specifically on comparisons between experimental spectra and photocurrent calculations as functions of photon energy and distinguish details of the 'surface' and 'bulk' electronic structures. Supported by the NSF (#DMR-9500654) and NATO (#CRG.940120).
10:20 am BREAK
10:40 am
ANISOTROPY OF SEGREGATION AT ANTIPHASE BOUNDARIES IN A BCC-BASED BINARY ALLOY: Qiang Wang, Long-Qing Chen, Department of Materials Science and Engineering, The Pennsylvania State University, University Park, PA 16802
A computer simulation on the kinetics and anisotropy of segregation at antiphase boundaries (APB's) in a model BCC-based binary alloy has been conducted using microscopic master equations in the first and second-neighbor pair approximations. Both stoichiometric and nonstoichiometric compositions are considered. It is shown that the degree of segregation at the APB's is highly anisotropic and the mobility of an APB at a given temperature is strongly composition-dependent. For the particular case of a cylindrical APB along the z direction, it is found that maximum segregation occurs along the [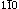 ]-direction and essentially zero along the [1 1 0]-direction. Despite the strong segregation anisotropy, however, it is demonstrated that the decrease in the radius of the cylindrical antiphase domain is linearly proportional to t1/2 where t is the time.
]-direction and essentially zero along the [1 1 0]-direction. Despite the strong segregation anisotropy, however, it is demonstrated that the decrease in the radius of the cylindrical antiphase domain is linearly proportional to t1/2 where t is the time.
11:10 am
SEGREGATION OF NIOBIUM SOLUTE IN NICKEL TOWARD GRAIN BOUNDARIES AND FREE SURFACES: Leonid Muratov, Bernard R. Cooper, Department of Physics, West Virginia University, Box 6315, Morgantown, WV 26506
The spatial redistribution of niobium atoms near the (100) and (111) free surfaces and selected grain boundaries (GB) of pure nickel has been considered in the low niobium concentration limit. Our simulations show a significant depletion in concentration of niobium immediately at a free surface. However, under the first two layers of pure nickel there exists a niobium enriched region with strongly temperature dependent concentration. This predicted non-monotonic distribution of niobium in the surface region may be very important for many applications and requires experimental confirmation. In contrast, at the grain boundaries, the concentration of niobium, pertinent to GB oxidation embrittlement, is predicted to be much higher than in the bulk. It monotonicaly decreases with the distance from GB, until reaching the bulk value. The calculation of the free energy uses atomistic potentials based on ab-initio quantum mechanical calculations, includes lattice relaxation around niobium atoms by using molecular dynamics (with 1440 or 2880 atoms in the modeling cell), and includes vibrational entropy phenomenologically within the local harmonic approximation.
11:40 am
EFFECTS OF OFF-STOICHIOMETRY ON ANTIPHASE BOUNDARIES IN MODEL Ll2 ALLOYS AND 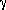 -TiAl PHASE: Patrick Tepesch, Mark Asta, Andrew Quong, Sandia National Laboratories, Livermore, CA 94550
-TiAl PHASE: Patrick Tepesch, Mark Asta, Andrew Quong, Sandia National Laboratories, Livermore, CA 94550
The effect of off-stoichiometry on the properties of conservative (100) and (111) antiphase boundaries (APB) in model L12 alloys are studied with the cluster variation method (CVM) in the Tetrahedron-Octahedron (T-O) approximation with first (V1) and second (V2) nearest neighbor pair interactions. Two model systems with  = V2/V1 = æ0.1 and æ1.0 are considered. The effect of off-stoichiometry on APB properties, such as the equilibrium () and non-equilibrium (°) antiphase boundary free energies, depends quantitatively and qualitatively on temperature and . First-principles results on the -TiAl phase show that these effects can be important in real systems. This work was supported by the U.S. Department of Energy, Office of Basic Energy Science, Division of Materials Science.
= V2/V1 = æ0.1 and æ1.0 are considered. The effect of off-stoichiometry on APB properties, such as the equilibrium () and non-equilibrium (°) antiphase boundary free energies, depends quantitatively and qualitatively on temperature and . First-principles results on the -TiAl phase show that these effects can be important in real systems. This work was supported by the U.S. Department of Energy, Office of Basic Energy Science, Division of Materials Science.
| Next Session | Technical Program Contents | ||
|---|---|---|---|
| Search | Materials Week '97 Page | TMS Meetings Page | TMS OnLine |